
In many ways, Siena's fifth birthday party last year was just like any other child's. There was cake, balloons, and – since it fell close to Cinco de Mayo – Mexican food, with piñatas for the children and margaritas for the adults. Siena's younger brother and younger cousin were there, alongside her parents, grandparents, and other family members.
What made the party unusual was that Siena wasn't expected to live past five years old. Just after her first birthday in 2017, she had been diagnosed with Tay-Sachs disease – a rare genetic disorder with no cure, no treatment, and no chance of survival for patients whose symptoms begin in early childhood.
But Siena is still alive, thanks in part to a new gene therapy that could revolutionise the treatment of Tay-Sachs. Developed by scientists at the University of Massachusetts (UMass), the therapy is still undergoing clinical trials and has only been tested on a handful of humans, of which Siena is one.
Both patients showed improvement, and Siena's mother Sara Margani told The Independent that her daughter is now more stable, without the seizures that she used to suffer, and with more control of her eye muscles. Other gene therapy programmes are also under way, raising the possibility that soon, for the first time in history, children diagnosed with Tay-Sachs might grow up to live long and full lives.
If so, it will be thanks to years of dedicated activism and fundraising by patients and the families of patients who have gathered money and pushed for research in the face of indifference from for-profit drug companies reluctant to fund research into a condition found in only one in 320,000 people in the US.
"Thirteen years ago, [there was] no hope on the horizon for any potential treatment for children who have been diagnosed," says Becky Benson, whose daughter Elliott died of Tay-Sachs in 2012. Now she works for the Allied Diseases Association (NTSAD), which helped fund the UMass research.
"I never believed that there was anything that could be done," she goes on. "I knew what we were facing... in today's world, it is a phenomenally amazing thing to think that there is hope – real, true, tangible hope – on the horizon for children who have been diagnosed with these diseases today."
'At that moment, all hope was lost'
The first systematic descriptions of Tay-Sachs disease came in the late 19th century. In 1881 Waren Tay, a British ophthalmologist working in London, began observing an odd red spot in some patients' retinas, later finding three cases in one family. In 1888, the American neurologist Bernard Sachs gave a more detailed account of the "arrested cerebral development" that followed, which left children "listless" and soon unable to move.
Both doctors observed the disease among Ashkenazi Jews, who for unknown reasons suffer much higher rates of Tay-Sachs (about one in 3,600). For years it was thought by some to be an exclusively Jewish condition, making it a useful prop in the "scientific" racism of the early 20th century. In fact, it can affect anyone and is also more common among French Canadians, some Amish people, and Cajun people.
Only in the Sixties did scientists discover why Tay-Sachs happens. Glitches in victims' genetic code leave them unable to produce an enzyme called HexA, which is crucial for breaking down a waste product of the brain called GM2 ganglioside. As GM2 builds up, it begins to damage and kill their brain cells. (There are other, similar disorders that stem from lack of sibling enzymes.)
For some people the genetic glitch is less severe, and GM2 builds up slowly enough that they are only diagnosed in adulthood. Their symptoms are difficult, but rarely fatal. For those whose symptoms are fast enough to surface in childhood, however, the prognosis has changed little since the 19th century.

For Ms Benson it began when Elliott was ten months old. Elliott (or, as Ms Benson habitually calls her, "Miss Elliott") was her second daughter, so she quickly knew that the child wasn't developing as she should, but doctors were sceptical and preferred to "wait and see".
An eye doctor thought she had a vision problem; an ear doctor thought she might be deaf. Then an ophthalmologist, like Tay, saw the red spot and was initially reluctant to explain what it was.
"That's when I knew we were in for something very serious," says Ms Benson, who lives in Washington state, US. "[Diagnosis] was just earth shattering for us, because I thought she needed physical therapy to get over the hump and be able to succeed... at that moment, for me, all hope was lost."
Sara Margani, 38, from Toronto in Canada, had a similar experience, starting with subtle developmental delays for Siena that led some to dismiss her as a "crazy mom". She says: "Devastated is really an understatement. You know, parents are given this diagnosis and they're basically told to take their kids home and love them and watch them as they decline."
The Marganis rolled up their sleeves and prepared to do whatever they could to help Siena. But: "Doctors looked at us like we were crazy. Like, 'what are you talking about? There's nothing to do.'".
Some palliative treatments are now available, such as cannabinoid-based anti-seizure drugs and a vibrating "shake vest" that dislodges fluid in the wearer's lungs or throat once they become unable to control the relevant muscles. But, according to NTSAD's director of family services Diana Pagonis, the basic picture remains the same as when she joined the group 27 years ago, when she knew nothing about Tay-Sachs and just needed a part time job.
"Thankfully," she says, "we've had those families who don't want to accept that."
Custom-made viruses open a back door to the brain
The same year that Siena was diagnosed, Miguel Sena Esteves and his colleagues were studying sheep.
To be specific, they were studying Jacob sheep, a strange black and white breed with up to six horns each, named for its resemblance to the Jacob's flock in the Book of Genesis. In 1999, a Texas farming couple had discovered signs of a mystery neurological disorder in their herd, which by 2010 had been confirmed as Tay-Sachs disease.
Instead of removing the affected animals, the farmers began selectively breeding more, and donated many of them to NTSAD and Auburn University to serve as a research population.
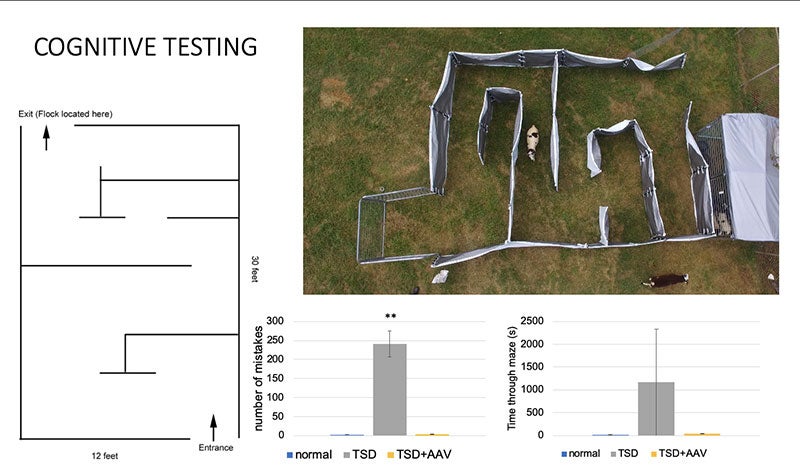
As a professor at the University of Massachusetts Chan medical School, Prof Esteves has been working for years to develop a treatment for Tay-Sachs. The challenge lies in a place that usually keeps our brains safe from harm: the blood-brain barrier, a special layer of cells that prevents anything in our blood from contaminating our central nervous system.
"I usually call the brain the prima donna of all the organs we have, because it likes to live in its own little environment," says Prof Esteves. "Nicely protected not only in the skull but also behind a physiological barrier that really does a bang-up job...
"If it wasn't for the blood-brain barrier, these diseases by now would be cured, or if not cured a major impact on them and our ability to manage them." Without the barrier, the missing chemical could simply be taken by patients as a drug, just as people with diabetes take regular insulin.
Certain types of viruses, though, are able to cross this barrier, providing a back door. What's more, viruses have evolved sophisticated mechanisms to latch onto human cells, take control of them, and insert a payload of genetic material that changes how they operate.
In deadly viruses such as SARS-Cov-2, which causes Covid-19, these infected cells are transformed into factories to produce more virus, according to the instructions of the genetic material that was inserted. If that payload is replaced with other genetic material – such as human DNA with the correct code to produce HexA or similar enzymes – then it effectively fixes the cell, allowing it to produce those enzymes normally.
A similar treatment using the same virus appears to be effective for spinal muscular atrophy (SMA), another often fatal disease stemming from lack of a crucial protein. The resulting drug, Zolgensma, has now been approved in at least 38 countries, and Prof Esteves says it has treated around 1,600 children.
And so, as scientists across the world raced to blunt SARS-Cov-2's ability to dodge our immune systems and penetrate our cells, Prof Esteves and his team have been working to make their harmless virus better at doing so.
Unlike other viruses, it is incapable of reproducing itself, so billions or even trillions of copies must be inserted in order to hit all the necessary cells. Since only a minuscule fraction get past the immune system, even small improvements in the virus's design can make a big difference.
The problem was funding. Developing and manufacturing custom viruses is an "immensely expensive endeavour", Prof Esteves says, Medical companies are often loath to investment money in treatments that can only be sold to a handful of patients worldwide. According to fundraisers, the UMass research, while promising, was marooned for some time without the money to go forward.
That is where patients and parents came in.
‘Researchers are pathetically underfunded’
The people affected by Tay-Sachs were organising to cure it before the Tay-Sachs gene was even discovered. NTSAD was founded in Brooklyn, New York City in 1957, and when the cause was it quickly rallied to organise mass screenings of parents to see if they carried recessive versions of the gene. In 2002, NTSAD began disbursing seed grants for research into the disease, and have so far given out $4 million (£2.9m).
"The families raised money and gathered before they had hope," says the group's communications head Susan Keliher. "They didn't know how long it was going to take, and they just did it."
For Ms Benson the group was her "saving grace", something that allowed her to move past the hopelessness of Elliott's own case. Having been supported by other parents whose children had died, she was able to support the next wave, maintaining the push for fundraising and research that might give hope to other, future kids.
"For so long it was just parents like us who were banding together," Ms Benson says says. "You know, you go to the grocery store and they ask you if you want to round up for breast cancer awareness, and I always think: 'everyone's aware!' Of course that's something that we need to fund, but no one's heard of Tay-Sachs." Indeed, she says Elliott was the first child in Washington state to be diagnosed in 20 years.

In the UK, Dan Lewi from Hither Green in south-east London set up the Cure and Action for Tay-Sachs Foundation (CATS) after his daughter Amelie was diagnosed at 15 months old. A network of European charities followed, in Spain, Portugal, and Germany.
"The thing you're normally told with a rare disease is, you'll never meet another family," Mr Lewi tells The Independent. "By working together, we've actually been able to show [companies] that patients do exist, and that we'd be an attractive option for any industry looking at developing a clinical study."
And in Canada, Sara Margani and her parents set up the Blu Genes Foundation in the aftermath of Siena's diagnosis to fund gene therapy research. "We started looking into what clinical trials and what therapies or research were available, and what we learned is that there really wasn't much," she says. "There were maybe a handful of teams all around the world that were studying this disease.
"We learned that UMass had done incredible research over ten years that was effectively shelved, and that these [kinds of] research teams are operating on shoestring budgets – they're pathetically underfunded."
The amounts such organisations can offer are small in absolute terms, but they can be crucial to getting research to a stage where the heavy hitters – governments and corporations – are persuaded it might be viable.
In 2017, NTSAD gave UMass an $80,000 grant to study gene therapy on the Jacob sheep, and later Blue Genes was able to raise more than 900,000 Canadian dollars (£523,000) from around 250 individual and corporate donors to begin human clinical trials.
"Without the foundations absolutely nothing would have happened, no question about it," says Prof Esteves. "It was fundamental... when people make strides forward and start to get good results, other sources of funding such as the NIH [National Institutes of Health] in the US, or the research council in the UK, those doors really open."
'We're seeing a little bit of light now'
Siena had the modified viruses injected through her spine. She was one of the first two patients given a conservative dose of UMass's gene therapy after a blizzard of back-and-forth between the researchers and the US Federal Drug Administration (FDA) to hash out how the treatment attempt should work.
These first two children were not part of a formal clinical trial. Both were treated as part of what the FDA calls “expanded access”, sometimes termed “compassionate use”, in which patients with life threatening diseases and no other satisfactory options can access experimental treatments if the benefits outweigh the risks.
Ms Margani says her daughter was eligible in part because she would probably have been too old to participate in future clinical trials. That decision, she adds, had no bearing on Blu Genes’ choice to fund the research and was made by UMass with the FDA’s approval.
Since then, Siena has seemed a little more alert, and the continual rolling movement of her eyes – she cannot control where they look – has stopped, as have her seizures. Gene therapy was not the only factor, since she is also taking anti-seizure drugs, but Ms Margani suspects it is what helped Siena reach age five.
The research was promising enough it has now been licensed by Sio Gene Therapies, a clinical trial company focused entirely on neurodegenerative diseases, which is conducting further trials with more patients and stronger doses. At a conference in October, the company said those patients had seen improvements in key markers of the disease and that six out of seven had "no evidence of overt disease progression", with the results differing by dose.
There are other projects too, such as a gene therapy technique using the game-changing CRISPR gene editing technology that Blu Genes is funding at the Hospital for Sick Children in Canada. NTSAD is tracking four clinical trials to treat Tay-Sachs and its sibling Sandhoff disease, plus five other trials to do with other similar conditions. The group has also just hired its first research director – the first time such a post has ever been necessary.
Still unknown is how gene therapy treatments will fare in the longer term, and whether they will merely ease patients' suffering, treat their symptoms, or actually cure Tay-Sachs. It's also unclear whether it will ever be possible to reverse the course of Tay-Sachs that has already set in, or whether such treatments will always be preventative in nature.
"With Siena the goal was never to cure," says Ms Margani. "I think we knew that wouldn't be possible with someone whose disease is so advanced as hers. The talk was more about stabilising disease, improving symptoms. In that context ,the therapy certainly has achieved that for Siena."
The good news is that even a small change to patients’ fortunes could transform lives. The youngest Tay-Sachs patients – that is, the ones with the fastest buildup of GM2 – typically have less than 0.1 per cent of normal HexA activity in their brains. Yet adult patients still only have 2-4 per cent of normal activity, and 5-10 per cent means there may be no symptoms at all.
In other words, Prof Esteves says, even a treatment that restored only 2 per cent of normal enzyme activity would dramatically change the outlook for this disease.

For now Siena seems stable, and there is a chance she will end up living more years – something previously unimaginable for children with Tay-Sachs. Ms Margani is cautious, saying the research landscape is promising but still far away from a time when parents of Tay-Sachs kids will have actual options for treatment.
"When your child is diagnosed with a terminal disease, there's there's no light," says Mr Lewi. "And now we're actually shedding a little bit of light, and there could be something in the future, it's exciting to know that it's not just doom and gloom."
Early diagnosis will be critical
Even if the treatment works, there are big problems to solve. Prof Esteves says that evidence from other diseases and clinical trials suggests that in most cases, gene therapy will only be able to "freeze" neurological damage, not reverse it.
That will make early detection and diagnosis critical, and yet many patients and parents – including Ms Margani and Ms Benson – faced delays and misdiagnoses. For adult patients, the path to diagnosis can be eight or ten years long and littered with red herrings.
Meanwhile, Mr Lewi says that the small number of patients, the rapid course of the disease, and the short life of its juvenile patients makes recruiting for clinical trials difficult. Children can start off as eligible but swiftly become ineligible, or pass away, before the trial begins.
The answer to all these issues could lie in universal screening of newborn babies, but Mr Lewin says many national health authorities have been reluctant to fund this so far (although Israel instituted a free parent screening system for Ashkenazi Jews in 1978). "They don't want to provide newborn screening for a condition that has no treatment, but it's a catch-22, because industry needs newborn screening to identify patients [to test] the treatment," says Mr Lewin.
In Canada, NTSAD helped fund a pilot programme for Tay-Sachs detection in newborns in Quebec. "When therapy becomes available for Tay Sachs and Sandhoff, discovering the diseases as early as possible will be essential," the organisation says.
There is also the issue of cost. Zolgensma, the gene therapy for SMA, became the most expensive in US history when it was approved by the FDA in 2019 at $2.1 million per patient. That is so out of reach for many that its maker, Novartis, launched a global lottery offering a hundred free treatments per year. For perspective, about 60,000 children are estimated to be diagnosed with SMA per year.
Even in countries with state-funded healthcare, officials may balk at such prices. England's NHS only agreed to fund cerliponase alfa, a drug that treats the incurable Batten disease, weeks before families of patients were due to take it to court. Mr Lewi says two eligible children in the UK died in between the end of clinical trials and the drug itself becoming available.
Perhaps, in comparison with 1881 (or even 2011), these are good problems to have. Ms Benson says: "We are standing on the shoulders of Miss Elliott, and all the children and patients who have been diagnosed with Tay-Sachs, leading us towards those treatments. I know that she's still a part of that effort, and that it's a valuable part."
In fact, Ms Benson's other daughter, who was seven years old when Elliott died and is now 17, hopes to study genetics at university, inspired by her experience with NTSAD and her sister.
In Toronto, if luck holds, Ms Margani hopes to be celebrating Siena's sixth birthday this May. "I can't see, knock on wood, anything preventing that from happening," she says. "I feel a sense of relief when her birthday passes, because it's like, 'okay, phew, we got through another year'.
"Her birthdays are very bittersweet. I'm happy that she's here with us and we're able to celebrate her, but I know she's not going to be with us forever, so that nagging thought – will this be the last birthday that I get to celebrate? – is always in the back of my mind. That's why they're so important to us, I guess."







